
O gene transportador do filamento de ligação ao ATP (ABC), ABCA4 (ABCR), foi caracterizado em 1997 como o gene causador da doença de Stargardt autossômica recessiva (STGD1). Logo em seguida, vários outros fenótipos foram associados a mutações no ABCA4, que agora emergiram coletivamente como a causa mais frequente de fenótipos de degeneração da retina da herança mendeliana. O ABCA4 funciona como um importante transportador (ou “flipase”) de derivados da vitamina A no ciclo visual. Diversas maneiras de aliviar os efeitos da proteína ABCA4 defeituosa, que causam acúmulo de 11-cis e all-trans-retinal em fotorreceptores e lipofuscina no epitélio pigmentar da retina, têm sido propostas. Embora o ABCA4 tenha provado ser um alvo de pesquisa difícil, avanços substanciais por meio de estudos genéticos, funcionais e translacionais têm permitido grande evolução em aplicações terapêuticas para patologia associada ao ABCA4, que deve estar disponível para pacientes no futuro próximo. Aqui, resumimos o status das opções de tratamento baseadas em terapia gênica das doenças associadas ao ABCA4.
A terapia gênica somática mostrou sucesso em vários modelos animais de degeneração retiniana (recessiva) (Campochiaro 2002; Allocca et al. 2006). Mais bem conhecidos foram os relatos de que um vírus adeno-associado recombinante (rAAV) transportando Rpe65 de tipo selvagem (rAAV-Rpe65) melhorou a função visual no modelo de cegueira infantil de Briard (Acland et al. 2001; Narfstrom et al. 2003; Le Meur et al., 2007). Sucessos semelhantes foram relatados para o RPE65 e LRAT em modelos de rato com vetores AAV e lentivirais (Dejneka et al 2004; Batten et al 2005; Bemelmans et al 2006; Yanez-Munoz et al., 2006…).
A substituição direta de genes representa uma opção terapêutica atraente também para o tratamento de todas as doenças associadas ao ABCA4, incluindo a doença de Stargardt (STGD1) (Allikmets et al. 1997; Molday e Zhang 2010). Inserir um gene funcionando normalmente dentro dos fotorreceptores portadores de ABCA4 mutado via terapia gênica deve, portanto, ser considerado como uma possível “cura” para doenças associadas a ABCA4 porque:
(1) Todas as doenças associadas a ABCA4 são recessivas, de modo que a adição de um gene funcional poderia restaurar completamente função visual e,
(2) a degeneração das células da retina em todas as doenças associadas ao ABCA4 é relativamente retardada, permitindo uma janela de tempo razoável para a intervenção terapêutica.
Atualmente, são utilizadas duas abordagens principais para a inserção do material genético no olho: uma baseada em vetores virais como veículos de entrega e a outra incluindo métodos de inserção de genes não virais. Até agora, a identificação de um vetor eficiente para a injeção do gene ABCA4 à retina tem sido dificultada pelo grande tamanho da sequência codificadora ABCA4 (6,8 kb) e pela expressão do gene / proteína ABCA4 exclusivamente em fotorreceptores (RP ) células, exigindo, portanto, vetores com grande capacidade de carga e capazes de transduzir PRs de forma eficiente. A maioria dos veículos de entrega que aceitam grande capacidade de carga, tais como lentivírus, adenovírus e vetores não virais, tem sido sugeridos como não capazes de transduzir eficientemente PRs, pelo menos nos modelos de camundongos. Ao mesmo tempo, vetores com capacidade eficiente de transdução de PR, por exemplo aqueles baseados em vírus adenoassociados (AAVs), têm uma capacidade de encapsulamento limitada a cerca de 4,7 kb (Colella e Auricchio 2012; Lipinski et al. 2013) e, portanto, não ser capaz de envolver a grande sequência de codificação ABCA4 de ∼7 kb. Recentemente, esforços significativos têm sido direcionados tanto para o desenvolvimento de sistemas baseados em AAV, que são capazes de fornecer genes grandes e a identificação de vetores lentivirais e não-virais com maior tropismo, ou seja, crescimento, de PR. Aqui vamos rever o estado atual do desenvolvimento da terapia gênica ABCA4.
INSERÇÃO NÃO VIRAL DO GENE EM DOENÇAS ASSOCIADAS AO ABCA4
Os vetores não virais oferecem várias vantagens sobre as estratégias baseadas em vírus, incluindo:
(1) toxicidade reduzida do vetor,
(2) falta de resposta imune contra o vetor e possibilidade de readministrar o vetor,
(3) uma grande capacidade transgênica, e
(4) produção de grau clínico simples e relativamente barata (Charbel Issa e MacLaren 2012).
No entanto, embora os vírus tenham evoluído para otimizar a injeção do gene viral no núcleo da célula hospedeira humana, o DNA sem cápsula entregue por vetores não virais precisa superar várias barreiras a serem expressas, tais como:
(1) degradação extracelular e imunológica resposta mediada por sensor de moléculas de DNA de fita simples ou dupla, como receptores toll-like,
(2) degradação citoplasmática e
(3) passagem através da membrana nuclear durante a divisão celular, o que não é possível em células pós-mitóticas, como PRs.
Além disso, a presença de barreiras físicas no olho, como as membranas vítreas limitantes internas / externas, a matriz inter-PR e as altas concentrações de glicosaminoglicanos presentes em todo o olho que sequestra o DNA, limitam ainda mais o acesso celular (Lipinski et al., 2013). Como consequência, muitos estudos forneceram evidências da maior eficiência da entrega de genes da retina viral versus não viral (Andrieu-Soler et al. 2006). Em particular, a inserção de DNA sem cápsula via injeção subretiniana entre o PR e o epitélio pigmentar da retina (LPE) (Liang et al. 2000) é altamente ineficiente (Charbel Issa e MacLaren 2012). Portanto, métodos químicos ou físicos têm sido usados para aumentar a eficiência da entrega de genes de vetores não virais para a retina externa. Métodos químicos, como lipossomas, polímeros e nanopartículas compactadas, são baseados na conjugação do DNA com composto catiônico sintético ou natural que protege o DNA da degradação mediada por nucleases e permite a passagem através das membranas celulares via endocitose e, em alguns casos, captação mediada por receptores. Métodos físicos, como eletroporação ou iontoforese, geralmente usam um estímulo elétrico para permeabilizar temporariamente a membrana e permitir que o DNA atravesse as membranas celulares. Até hoje, no entanto, a evidência da inserção efetiva de genes nas células retinianas externas permanece escassa e o sucesso da maior parte da tecnologia de entrega não-viral tem sido limitado à camada de células EPR (Kachi et al. 2005; Johnson et al. 2008; Souied et al. al. 2008). Entretanto, recentemente, a nanopartícula de DNA compactado (NP) CK30-NP, à base de polilisina, mostrou melhora significativa na eficácia da transferência gênica ocular em comparação com os métodos não virais anteriores (Farjo et al. 2006). Nas nanopartículas compactadas, em particular, a molécula de DNA é compactada por peptídeos de lisina 30-mer substituídos com polietilenoglicol (PEG) (CK30PEG) (Liu et al. 2003). Seu diâmetro mínimo (geralmente 8–20 nm) é tal que permite entrar prontamente no núcleo (Liu et al. 2003) das células em divisão e não-divididas, permanecendo epissomal. É importante ressaltar que essas partículas não têm limitações teóricas de envolvimento de DNA e foram testadas com sucesso com plasmídeos de até 20 kb de comprimento (Fink et al. 2006). Notavelmente, a administração sub-retiniana de CK30-NPs resulta em transdução de PR extensiva, embora com alguns padrões inexplicáveis de expressão de transgenes (Kumar-Singh 2008) na ausência de respostas imunes detectáveis ou toxicidade (Farjo et al. 2006; Ding et al. 2009). Além disso, em contraste com outras abordagens não-virais, as NP-CK30 dirigem a expressão gênica a longo prazo após a inserção subretiniana no olho do rato (Conley e Naash 2010). Dado o sucesso da CK30-NPs mediar o resgate fenotípico em modelos de roedores de retinose pigmentar (Cai et al. 2009, 2010) e seu perfil de segurança e eficácia favorável em um ensaio clínico em humanos para fibrose cística (Konstan et al. 2004). Han et al. (2012) testaram recentemente CK30-NPs para entrega de ABCA4 no modelo de rato Abca4 – / – da doença de Stargardt. Este modelo de rato mostra algumas das características clínicas associadas ao STGD1, tais como o acúmulo de lipofuscina (A2E) no EPR (Weng et al. 1999; Mata et al. 2001), espessamento das células do EPR (Allocca et al. 2008; Radu et al., 2008; Conley et al., 2012), recuperação tardia da dessensibilização leve (Weng et al. 1999; Maiti et al. 2006; Allocca et al. 2008; Radu et al. 2008) e desbaste do nuclear externo ( Células PR em camundongos albinos KO (Radu et al. 2008; Wu et al. 2010a). Neste modelo de ratinho, após injeção sub-retiniana das NP-CK30 portadoras do gene ABCA4 humano, Han et ai. (2012) detectaram a expressão do transgene ABCA4 por até 8 meses após a injeção e encontraram melhora na recuperação da adaptação ao escuro e redução do acúmulo de lipofuscina. Esses dados promissores forneceram a primeira evidência de entrega mediada por vetores não-virais efetiva de genes grandes, como ABCA4, para a camada de células EPR.
INJEÇÃO AAV DO GENE NAS DOENÇAS ASSOCIADAS AO ABCA4
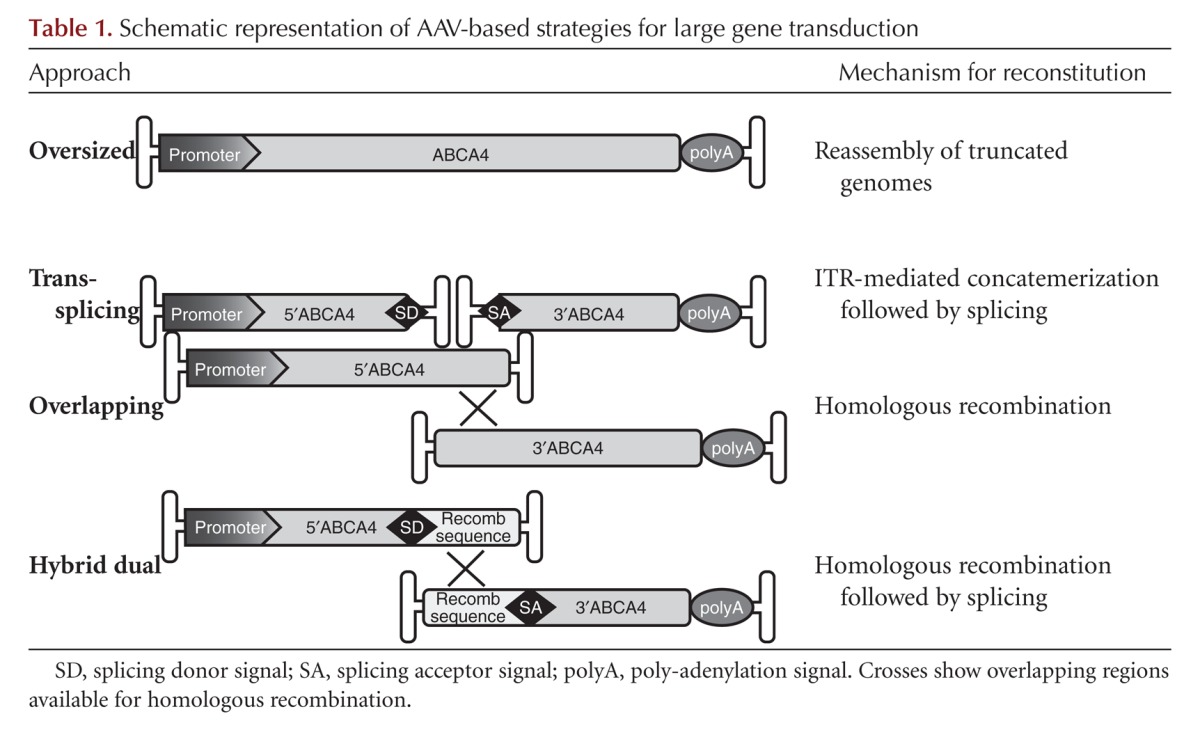
Os vetores derivados de AAV são atualmente os veículos mais favorecidos para a inserção de genes terapêuticos para a retina, porque têm baixa imunogenicidade, perfil de segurança favorável e suportam a expressão de transgene a longo prazo após uma única administração (Colella e Auricchio 2012; Vandenberghe e Auricchio 2012). O AAV �um v�us (ah?) pequeno sem membrana de 25 nm que envolve um genoma de DNA de cadeia simples linear de ~ 4,7 kb. Um dos pontos fortes da plataforma vetorial AAV é a disponibilidade de mais de 100 formas diferentes chamadas de sorotipos AAV, que são isolados como clones infecciosos ou moleculares, e que diferem em suas proteínas de capsídeos da superfície externa. Os capsídeos podem ser facilmente trocados entre vários AAVs para gerar vetores híbridos que contêm um genoma com as mesmas repetições terminais invertidas de AAV (ITRs) e o capsídeo de um sorotipo diferente (Auricchio 2003). AAVs obtidos através deste sistema de transcapsidação são denominados AAV2 / n, onde o primeiro número se refere às ITRs e o segundo ao capsídeo (Surace e Auricchio 2008). Cápsulas diferentes conferem características diferentes de tropismo e transdução. Os vetores iniciais de AAV foram baseados no sorotipo AAV 2 (AAV2 / 2), o sorotipo mais prevalente em humanos. No entanto, o AAV2 / 2, embora seja um excelente vetor de transferência de genes para a transdução de RPE ou células ganglionares da retina, é relativamente ineficiente na transdução de outros tipos de células da retina, tais como PR. Como a maioria das mutações que causam degeneração retiniana hereditária, incluindo STGD1, ocorre em genes expressos em PRs (Vandenberghe e Auricchio, 2012), essa preocupação levou à busca de sorotipos de AAV capazes de superar essa limitação. Os vetores AAV2 / 5, 2/7, 2/8 e 2/9 têm todos os PRs eficientemente transduzidos, além do RPE (Auricchio et al. 2001; Lotery et al. 2003; Allocca et al. 2007), com o AAV2 / 8 sendo o sorotipo mais eficiente em camundongos (Auricchio et al. 2001; Lotery et al. 2003; Allocca et al. 2007), suínos (Mussolino et al. 2011), cães (Stieger et al. 2008) e primatas não humanos (Vandenberghe et al. 2011). A principal limitação para o uso de AAVs para a substituição de genes continua sendo sua capacidade de acondicionamento, que é considerada restrita ao tamanho do genoma parental (4,7 kb), e assim dificulta o tratamento de certas formas de doenças retinianas hereditárias causadas por mutações em genes em que o DNAc excede 5 kb, tal como ABCA4. Assim, diferentes estratégias para superar as limitações de carga do AAV foram investigadas. Uma é baseada em embalagens de genomas superdimensionados, isto é, maiores que 5 kb (Grieger e Samulski 2005; Allocca et al. 2008; Hirsch et al. 2010). Notavelmente, os AAVs superdimensionados têm sido usados com sucesso para inserir ABCA4 na PR de camundongos com Abca4 mutado- resultando em melhoria morfológica e funcional significativa e estável da retina (Allocca et al. 2008). No entanto, o mecanismo subjacente à transdução de PR mediado pelo AAV permanece indefinido, pois os genomas contidos em vetores de AAV superdimensionados parecem altamente heterogêneos em tamanho e são predominantemente mais curtos que o esperado (Dong et al., 2010; Hirsch et al., 2010; Lai et al., 2010 Wu et al., 2010b; Hirsch et al., 2013), o que limita seu uso no cenário clínico.
Fonte: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4448589/
Para compreender o significado dos termos científicos deste texto, consulte nosso Dicionário, clicando AQUI.